金子研の論文が Journal of Chemical Information and Modeling に掲載されましたので、ご紹介します。タイトルは
です。これは学部卒の根本康平さんが四年生のときに取り組んだ研究の成果であり、物性・活性・特性の目的変数 y の目標値を入力すると、それを達成する分子の化学構造を直接的に出力する数理モデルに関する論文です。イメージとしては、下の図のように、化学空間の中から本当に欲しい (未知の) 分子だけをスッと取ってくる感じです。

分子設計・材料設計において、分子の化学構造から変換された分子記述子やフィンガープリントなどの特徴量 x と分子・材料の物性・活性・特性といった目的変数 y との間で数理モデル y = f(x) を構築します。モデルを構築したら、新たな分子に対して、分子の化学構造から変換した x の値から y の値を予測したり、y の目標値を達成するための分子の化学構造を設計したりします。
分子設計に必要なことは、y の目標値からそれを実現するための分子を導くことです。すなわち、数理モデルの逆解析です。ただ、一般的なモデルの逆解析で行われていることは、分子の化学構造をコンピュータで大量に生成し、それらを x に変換した後にモデルに入力して y の値を予測し、予測値が良好な分子を選択する、すなわち順解析を網羅的に繰り返す擬似的な逆解析にすぎません。低分子で、原子の種類をある程度しぼっても、10 の 60 乗以上の化学構造が存在するといわれており、そもそも全ての分子を生成することは不可能です。そのため、ある狭い範囲で分子を生成して、それらの y を予測することにすぎず、網羅的に有望な分子を探索することはできません。
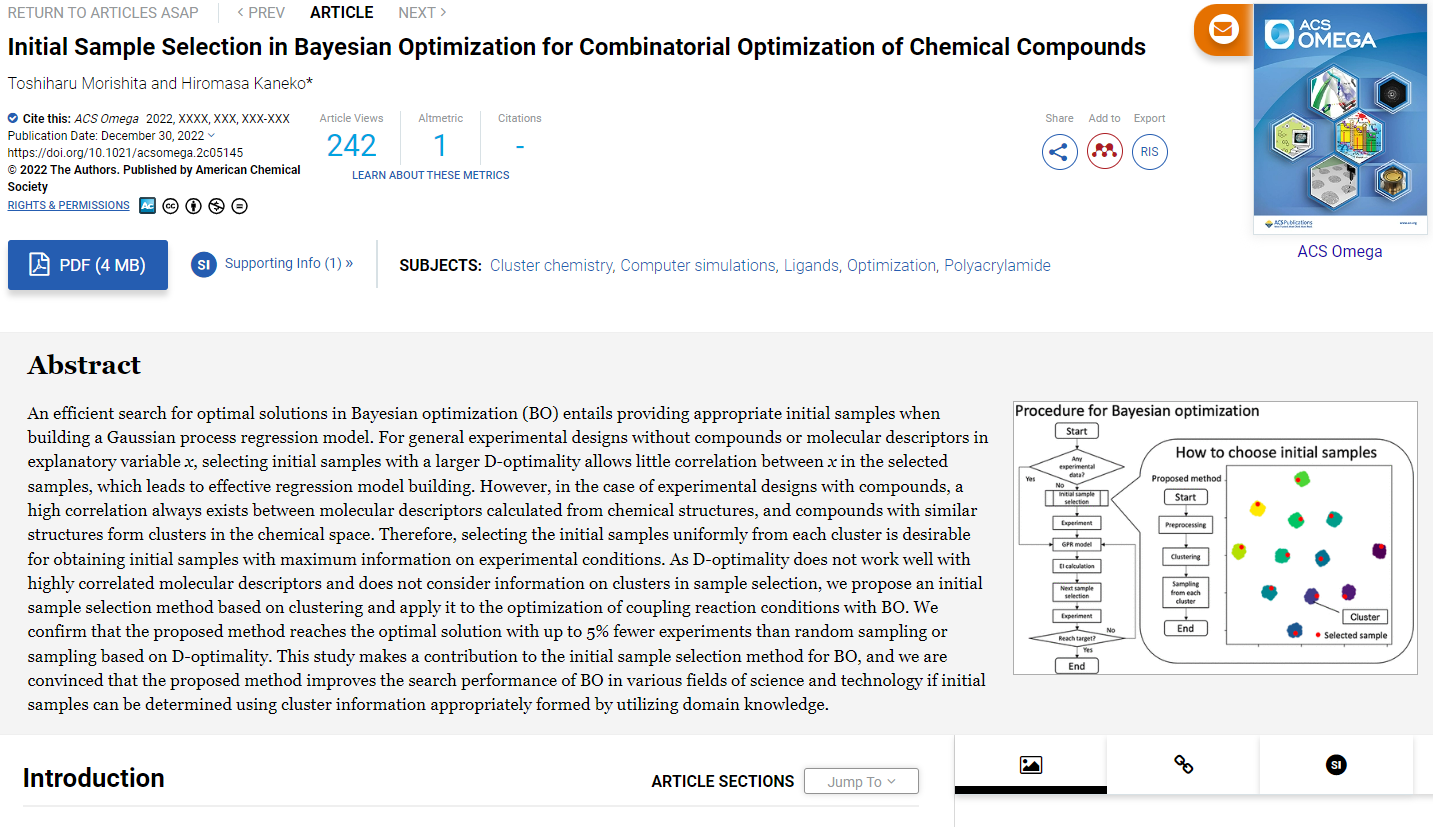
そこで本研究では物性や活性の y が目標値を持つ分子を直接的に生成するため、Chemical Variational Auto-Encoder (VAE) と Gaussian Mixture Regression (GMR) を組み合わせた手法de novo direct inverse QSPR/QSAR を開発しました。まず、下の図のように Chemical VAE モデルと GMR モデルを構築します。
分子を用いて学習させた Chemical VAE モデルを用いることで、分子の SMILES から潜在変数 x に変換したり、x から SMILES に変換したりできます。分子の物性や活性 y や Chemical VAE モデルにより変換された潜在変数 x を用いた学習させた GMR モデルを用いることで、x から y を予測したり、y から x を予測したりできます。
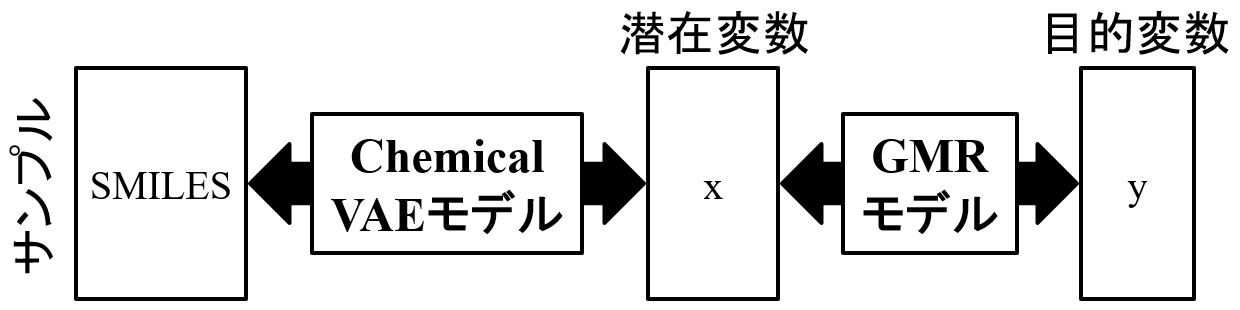
これらの Chemical VAE モデルと GMR モデルを用いて、まず y の目標値を GMR モデルに入力することで Chemical VAE の潜在変数 x の値を出力し、次にその x の値を Chemical VAE モデルに入力することで SMIELS もしくは分子を出力します。
さらにはそれらの複数の物性が目標値を持つような構造生成を行ったところ、従来のchemical VAEでは構造生成できない場合でも、提案手法により目標の範囲内に効果的に分子を生成できることを確認した。
いくつかの物性を対象にした分子設計を行ったところ、提案手法を用いることで、従来手法より効果的に分子が生成できること、そして所望の y の範囲に入る分子を適切に生成できることを確認しました。さらに、y が複数の物性の場合でも、提案手法により各 y が目標値に近い分子を生成できました。
興味のある方は、ぜひ論文をご覧いただければと思います。どうぞよろしくお願いいたします。
以上です。
質問やコメントなどありましたら、twitter, facebook, メールなどでご連絡いただけるとうれしいです。