分子設計、つまり目的に応じた化学構造の設計についての話です。分子設計を応用する対象が、医薬品のときは、医薬品設計や薬物設計と呼ばれたりもします。
分子設計とは?
分子設計の目的は、高い機能をもつ化合物をつくることです。たとえば、
- よく効く薬 (となる化合物)
- 収率が高くなる触媒
- やわらかくて強度の高いゴム
などです。このような高機能性化合物をつくるためには、どのような化学構造にすればよいか設計するのが、分子設計です。
下で丁寧に説明します。
化学構造と化合物
ここでは、「化学構造」と「化合物」とを使い分けます。化学構造とは、水素原子・炭素原子・酸素原子などの原子で構成される構造です。それぞれの原子がどこに位置するかと、各原子間はどのような結合か、で表されます。下図が化学構造の例です。
一方、「化合物」は実際に存在する分子です。たとえば水・エタノール・ベンゼンなど、実際にありますよね。もちろん、化合物にも化学構造があります。
「化学構造」とだけいったときは、実際に物質として存在してもしていなくても構いません。紙の上に描いただけの「化学構造」かもしれませんし、パソコンで描画した「化学構造」かもしれません。
「化合物」はしばしば、沸点・融点・水溶解度などの物性の値や、タンパク質に対する活性の値などが測定されています。何らかの目的があって化合物を合成したわけですので、少なくともその目的に対応する物性・活性は測定されるはずです。たとえば薬の元となる化合物を作ろうとするとき、目的のタンパク質に対する活性の値を測定して、薬になりうるかどうか、判断しなければなりませんよね。
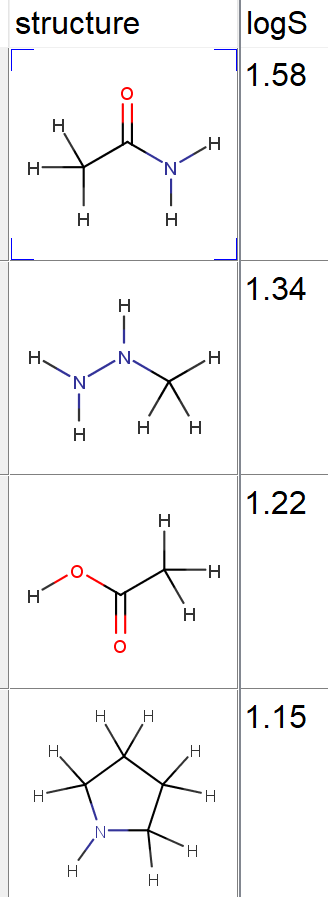
物性値もしくは活性値が測定されている化合物群は、下の図のように整理できます。物性は水溶解度Sであり、その対数の値が格納されています。
物性推定モデル・活性推定モデル
分子設計で大事なのは、ある化学構造を合成して得られる化合物の物性値もしくは活性値を、合成せずに推定することです。化学構造から物性値・活性値を推定するモデルを、それぞれ物性推定モデル・活性推定モデル、と呼びます。このモデル構築に関する研究分野が、定量的構造物性相関 (Quantitative Structure-Property Relationship, QSPR)・定量的構造活性相関 (Quantitative Structure-Activity Relationship, QSPR) です。
物性推定モデル・活性推定モデルをつくり、活用する流れは以下の図のとおりです。
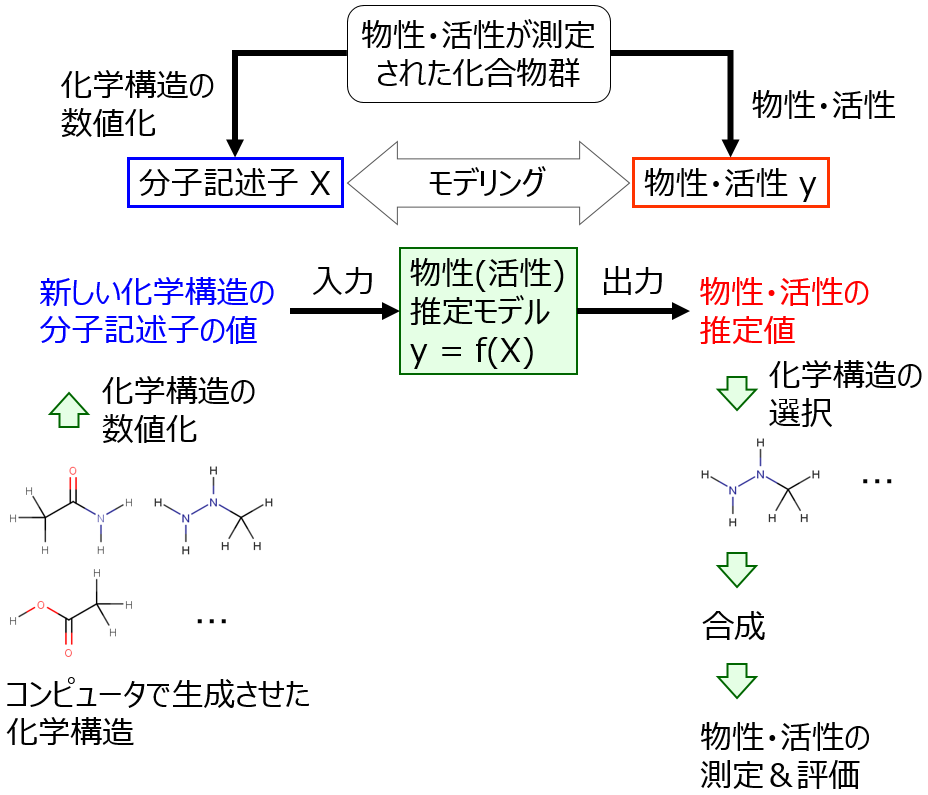
この流れにそって順に説明します。
なお、そもそもの前提は、推定したい物性・活性について、すでに物性値・活性値が測定されている化合物群 (化合物データベース) が存在していることです。
化学構造の数値化
まず、化合物の化学構造を数値化します。化学構造が与えられたときに、それを特徴づける量を計算するわけです。たとえば、分子量が分かりやすいと思います。もちろん分子量だけでなく、炭素原子の数・環の数・ファンデルワールス体積など、化学構造を数値化するいろいろな量があります。これらを、分子記述子 (molecular descriptor) や構造記述子 (structure descriptor) と呼びます。
たとえば、Dragon というソフトウェアを使うと、4885個もの記述子を計算できます (2017年10月22日の情報)。Codessa を使うと量子化学的な特徴を考慮した記述子が計算されます。PythonやC++を使う人は、RDKitで記述子を計算すると便利です。
(Molecular) fingerprint という、化学構造における部分構造などの構造的特徴が存在するかしないかを、それぞれ1, 0 で表現する記述子もあります。RDKitで計算できる fingerprint はこちら。
分子の3次元構造に基づいて、3次元座標から記述子を計算する方法もあります。ドッキングシミュレーションや分子動力学シミュレーションなどのシミュレーションによって記述子計算することも考えられます。
目的の物性・活性を推定するために適した記述子を開発することも、研究の方向性としてあります。
回帰分析・クラス分類 (モデリング)
物性や活性が測定された化合物群において、化学構造を数値化した分子記述子を計算できたとします。その後、構造記述子と物性・活性との間で回帰分析を行います。もし、物性・活性にあたるものが、連続値ではなくラベルで与えられるときは (薬か薬でないか、など)、クラス分類になります。つまり、物性推定モデルや活性推定モデルは、回帰モデルやクラス分類モデルであったわけです。回帰分析やクラス分類の手法についてはこちらをご覧ください。ここでの回帰分析やクラス分類のことを、一般的にモデリングと呼びます。
適切に分子設計を行うためには推定精度の高いモデルが必要です。つまり、新しい化学構造に対して、推定された物性・活性の値と、実際の物性・活性の値との誤差が小さくなくてはならないわけです。そのため、モデルの推定精度を向上させることを目的として、いろいろな研究が行われています。
また、構築されたモデルをどのように利用するか、たとえば重要な記述子はどれか、モデルがどんな意味をもっているか、についても興味をもつ人がいます。
モデルの適用領域・適用範囲
もちろん推定性能も重要ですが、回帰モデル・クラス分類モデルを利用するためには、モデルの適用領域・適用範囲 (Applicability domain, AD) を考えなければなりません。モデルの適用範囲・適用領域をどのように設定するかや、どのように利用するか、についても1つの研究の方向性です。
物性推定モデル・活性推定モデルの活用、分子設計
物性推定モデル・活性推定モデル (回帰モデルやクラス分類モデル) があれば、新たな化学構造を数値化してモデルに入力することで、実際にその化学構造の化合物を合成して物性・活性を測定することなく、物性・活性の値を推定することができます。なので、こんなことができます。
- 大量に化学構造を生成する
- 生成した化学構造の記述子を計算する
- 記述子の値をモデルに入力して物性・活性の値を推定する
- 推定値が望ましい値になった化学構造だけを選択する
- 選択した化学構造を実際に合成して、物性・活性を測定・評価する
これによって、効率的に目標の物性・活性の値を達成する化合物を得ることができると考えられます。これは、最もシンプルな分子設計の流れです。既存の化合物データベースを用いて、物性推定モデル(活性推定モデル)を作り、大量の化学構造の中から目標の物性値 (活性値) を満たすような化学構造を選ぶことで、望ましい化学構造を設計したことになります。
シンプルな分子設計の問題点と解決策の例
問題:存在する可能性のある化学構造が膨大にある
上では、物性推定モデル・活性推定モデル (回帰モデルやクラス分類モデル) を化学構造の “フィルター” として用いる話をしました。つまり、大量に化学構造を生成しておき、その中から “フィルター” を通った化学構造のみを選択する、というわけです。
しかし、このアプローチには限界があります。こちらで示されているように、たとえば低分子の有機分子の化学構造の候補だけでも、10の60乗 (1060) 個を超える候補があります。これらすべてをコンピュータで扱うことは、現実的に無理です。考えうる網羅的な化学構造の中から、目標の物性値・活性値をもつような化学構造を探したくても、できないわけです。
そこで、目標の物性値・活性値をもつ構造のみ扱おう、という話になります。たとえば、遺伝的アルゴリズムを用いて目標の物性値・活性値を持つような化学構造を生成するようなアプローチだったり、ベイズの定理を利用して目標の物性値・活性値をもつ化学構造が存在する可能性の高い記述子の範囲を設定して そこに集中的に化学構造を生成するアプローチだったりです。
もちろん、モデルの適用範囲・適用領域を考慮して化学構造を設計する必要もあります。
ただ、これらのような目標の物性値・活性値を満たすような化学構造を集中的に生成する手法にも問題があります。たとえば、
- 生成された化学構造が安定的に存在するか不明
- 化学構造を実際に合成できるかどうか不明
- どのように合成したらよいか不明
といったことです。
問題:化合物として作れるかについて考慮されていない
適当に化学構造を作ったのでは、
- その化学構造を実際に合成できるのか?
- どのように合成すればよいのか?
が わかりません。
そこで、化学反応のデータベースに基づいて化学構造を生成する方法もあります。いろいろな化学反応の反応中心をデータベースとして保存しておき、化学構造をその反応中心に基づいて仮想的に反応させすことで、新たな化学構造を作るわけです。もちろん、それで100%反応が進むわけではありませんが、合成可能性や合成経路まで考慮できるわけです。
ただこの方法にも問題点があります。たとえば、
- 生成された化学構造が安定的に存在するか不明
- 目標の物性値・活性値を満たす化学構造のみを扱うアプローチではないため、網羅的に多様な化学構造を探索することは困難
です。
問題:複数の物性・活性を考慮しなければならない
分子設計をするときの問題点として、1つの物性・活性だけ考慮すればよいわけではない、ことがあげられます。たとえば、医薬品を設計するとき、あるタンパク質に対する活性だけでなく、他のタンパク質に対する活性、つまり副作用、以下のADMETを考慮しなければなりません。
- Absorption (吸収)
- Distribution (分布)
- Metabolism (代謝)
- Excretion (排泄)
- Toxicity (毒性)
この問題に対して、物性推定モデル・活性推定モデル (回帰モデルやクラス分類モデル)を必要な数だけ増やす、というアプローチがあります。
ただ、このアプローチにも、たとえば
- 生成された化学構造が安定的に存在するか不明
- 化学構造を実際に合成できるかどうか不明
- どのように合成したらよいか不明
といった問題があります。
研究の方向性 まとめ
化合物データベースに基づく分子設計・化学構造設計の研究の方向性をまとめます。
- 目的に応じた分子記述子の設計
- 推定性能の高いモデルを構築できる手法の開発
- モデルの適用領域・適用範囲の設定
- モデルの利活用もしくはモデルの解釈
- 目標の物性値・活性値を満たすための網羅的な化学構造の生成
- 化合物としての存在可能性を考慮した化学構造の生成
- 合成可能性・合成経路を踏まえた化学構造の生成
- 複数の物性・活性を考慮した化学構造の生成
以上です。
質問やコメントなどありましたら、twitter, facebook, メールなどでご連絡いただけるとうれしいです。