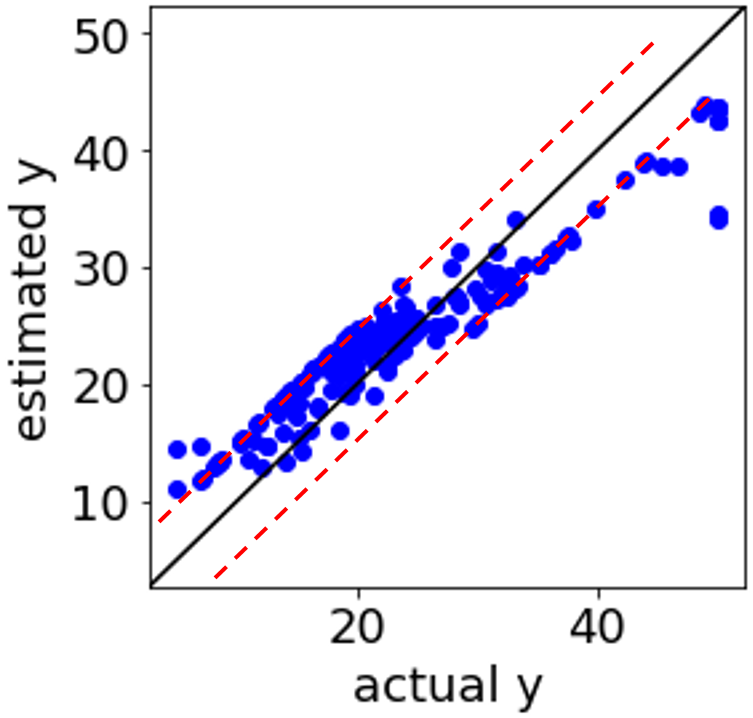
データセットがあると、データセットの可視化・クラスタリング・クラス分類・回帰分析などができるようになったり、モデルの適用範囲を設定したり、実験計画法により実験候補を選択できます。こちらにいろいろな手法の説明があります。

ただ、どの手法を使うにしても、サンプルが数値の変数 (特徴量・記述子など) で表されていなければなりません。化合物をサンプルとしてデータ解析や機械学習を行うとき、化学構造の情報を数値化する必要があります。
まずは、数値化しやすい形式で化学構造を表現する方法について説明します。実際はいろいろな方法がありますが、今回は代表的な 2 つの方法を扱います。
SMILES
1 つは Simplified Molecular Input Line Entry System (SMILES, スマイルズ) です。SMILES により、一行の文字列で化学構造が表現されます。例えばエタノールの SMILES は CCO です。水素原子は省略されます。下図のアラニンのように化学構造に分岐があると CC(N)C(=O)O のように () を用います。
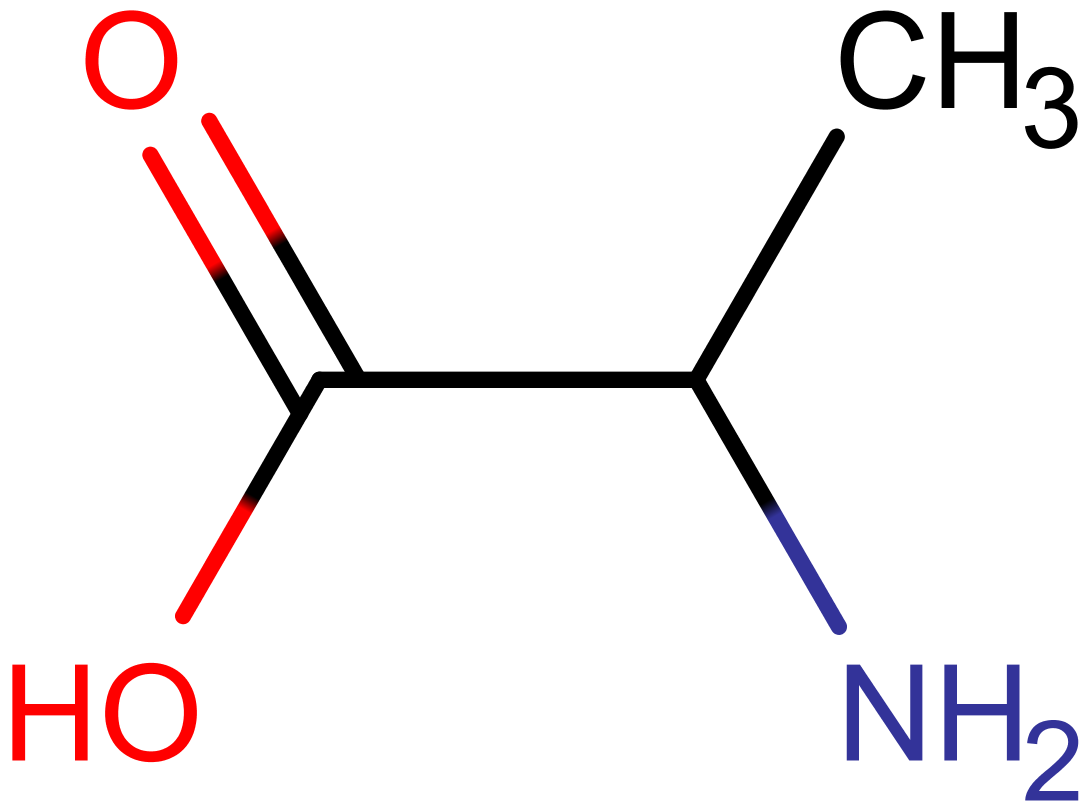
二重結合は =、三重結合は # です。SMILES では芳香族環の原子は小文字になり、例えばベンゼンは c1ccccc1 です。同じ数字で環の始まりと終わりを表します。他の例では、
- シクロヘキサン: C1CCCCC1
- トランス-2-ブテン: C/C=C/C
- シス-2-ブテン: C/C=C\C
などがあります。その他の SMILES の規則については英語版Wikipedia が参考になります。ただし、こちらにある手法で解析するだけでは、構造式から SMILES を書いたり SMILES から構造式を書いたりすることは基本的にないため、規則を覚える必要はありません。ChemDraw JS や Marvin JS などのウェブサービスや Marvin の MarvinSketch などの構造式描画ソフトウェアを用いることで、分子の構造式を描き、それを SMILES に変換したり、SMILES から構造式にして表示したりできます。
SMILES ではシス-トランス異性体などの立体配置による異性体の違いは判別できる一方で、シクロヘキサン環のいす形・ねじれ舟形などの立体配座による異性体の違いは表現できません。注意しましょう。SMILES では一つの分子が一行で表されるため、化学構造のファイルのサイズを抑えられます。多くの分子を扱うときに有効です。
MOL file
化学構造のもう一つの表現方法は MOL file (モルファイル) です。MOL file には原子の種類やその位置に関する情報、原子間の結合の情報等が含まれています。(さきほど上で図で示した) アラニンに対応する MOL file を下図に示します。

ChemDraw JS や Marvin JS や Marvin の MarvinSketch などで化学構造を描画すれば、MOL file 形式で保存できます。それをサクラエディタや mi などのテキストエディタで開くと、上の図と同様に表示できます。
MOL file の最初の三行は header block と呼ばれ、MOL file を作成したソフトウェアの名前や分子の名前など、MOL file を特定するための情報が記載されます。三行分あれば何も記載されていなくても問題ありません。
次の行は counts line と呼ばれ、原子数と結合数が記載されます。アラニンは原子数 13、結合数 12 であることが分かります。
次の行から原子数分の行はatom block と呼ばれ、各原子の三次元座標と種類が記載されます。例えば1番目の原子の三次元座標は (1.7168, 0.1992, 0) であり種類は炭素原子 C です。次の行から結合数分の行は bond block と呼ばれ、2 つの原子の番号とその間の結合の種類が記載されます。例えば最初の行からは、atom block における 1 番目の原子 ((1.7168, 0.1992, 0) の C) と 2 番目の原子((0.2341, 0.4264, 0) の C) とが単結合 (1) で結合していることが分かります。なお結合の種類として、2 は二重結合、3 は三重結合を表します。最後の行の M END は一つの分子の終わりを意味します。
その他、0 が並んでいる部分などの詳細を知りたい方は MOL file の仕様書をご覧ください。ただしMOL file を直接作成することはありません。SMILES と同様にして、ウェブサービスやソフトウェアを利用して、描画した構造式から MOL file に変換したり、MOL file から構造式として表示したりします。また ChEMBL (ケンブル) や PubChem (パブケム) などの公共のデータベースから MOL file をダウンロードすることも可能です。
上図のアラニンの MOL file では水素原子の情報が明示的に記載されていますが、ファイルの大きさを小さくするために水素原子を省略した下図のような MOL file も使用できます。

MOL file は SMILES よりファイルサイズが大きくなりますが、SMILES で区別できなかった立体配座による異性体の違いを、MOL file であれば区別できます。
化合物群のデータセットの扱い
こちらのプログラミング課題において回帰分析等で用いる水溶解度のデータセットは、水溶解度が測定された 1290 個の化合物です。このデータセットでは、化合物の化学構造がすでに数値化されているファイルもありますが、元は化合物をサンプルとして、各化合物の化学構造情報と物性情報で構成されています。このような化合物のデータベースを整理する方法や扱う方法について説明します。
データベースの整理法の一つは、こちらの csv ファイルのように化合物ごとに SMILES と物性・活性等を併記する方法です。csv ファイルを見ると、分子ごとに SMILES と水溶解度の値があります。物性・活性はなくても、また複数あっても構いません。
もう一つの整理法は、こちらのファイルのような Structure-Data File (SDF) と呼ばれるデータベース形式で整理する方法です。SDF のファイルの拡張子が sdf であることから、(F は file を意味しますが) SDF ファイルとも呼ばれます。公共のデータベースから化合物群をダウンロードするときは SDF であることが多いです。sdf ファイルをテキストエディタで開くと、MOL file が並んでいることが分かります。各化合物において、MOL file に続いて 「> <プロパティ名> (任意の文字列) 」 の後にプロパティの情報があります。今回のプロパティは Boiling Point です。プロパティの種類は複数でも構いません。一つの化合物の終わりは $$$$ で表現されます。
化学構造の数値化
SMILES や MOL file で表現された化学構造を読み込み、それに基づいて、化学構造を数値化します。数値化した特徴量のことを、分子記述子や構造記述子、もしくは単に記述子と呼びます。こちらのプログラミング課題では RDKit で記述子を計算する課題があります。他にも、mordred や PaDEL や DRAGON などでも計算できます。不明な記述子については、RDKit であれば記述子リストにある論文を参照するとよいでしょう。一般的な記述子に関しては本で辞書的に調べるとよいです。
また化学構造を数値化する方法の一つに、下の図のようにして化学構造の構造的な特徴に基づいて、化学構造を 0, 1 のビット列であらわす fingerprint (フィンガープリント) もあります。
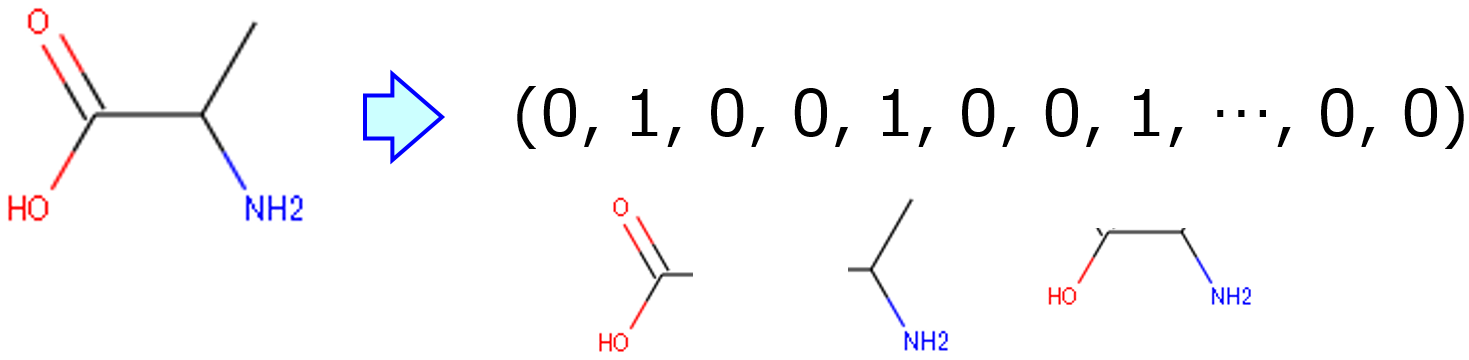
フィンガープリントは主に化学構造間の類似度の計算に用いられます。たとえば tanimoto 係数もしくは Jaccard 係数により化学構造の間の類似度を定量的に評価できます。
以上により化合物群において記述子のデータセットが得られます。データの可視化・クラスタリングをしたり、実験計画法により化学構造を選択したり、目的変数の情報も用いてクラス分類や回帰分析をしたりできるようになります。
化合物のデータセットを扱うときの注意点
例えばクラス分類や回帰分析を行うとき、データセットをトレーニングデータとテストデータに分けた後、記述子の標準化 (オートスケーリング) をします。記述子をその標準偏差で割りますが、すべての化合物で値が同じで標準偏差が 0 の記述子があると、0 で割ることになり計算できません。事前に標準偏差が 0 の記述子を削除する必要があります。削除後、適切にオートスケーリングできるようになります。なお、テストデータで削除する記述子をトレーニングデータで削除した記述子と揃えるため、テストデータではなくトレーニングデータにおいて標準偏差が 0 の記述子を削除することに注意してください。
標準偏差が 0 の記述子の削除は、トレーニングデータとテストデータに分割した後に行ってください。分割前に削除しても、分割後に新たに標準偏差が 0 になる記述子が出てくる可能性があるためです。
以上です。
質問やコメントなどありましたら、twitter, facebook, メールなどでご連絡いただけるとうれしいです。