
これから書くことか過去の失敗例です。ご注意ください。
回帰モデルでもクラス分類モデルでも、あるデータセットに基づいてモデルが構築されたとします。そのモデルを用いて新しいサンプルの目的変数 y の値を推定するとき、新しいサンプルがモデル構築用データセットのサンプルに類似していないと、y の推定値の誤差が大きくなってしまいます。モデルが、本来の推定性能を発揮できるデータ領域のことを、モデルの適用範囲・モデルの適用領域 (Applicability Domain, AD) とよびます。AD の詳細についてはこちら。
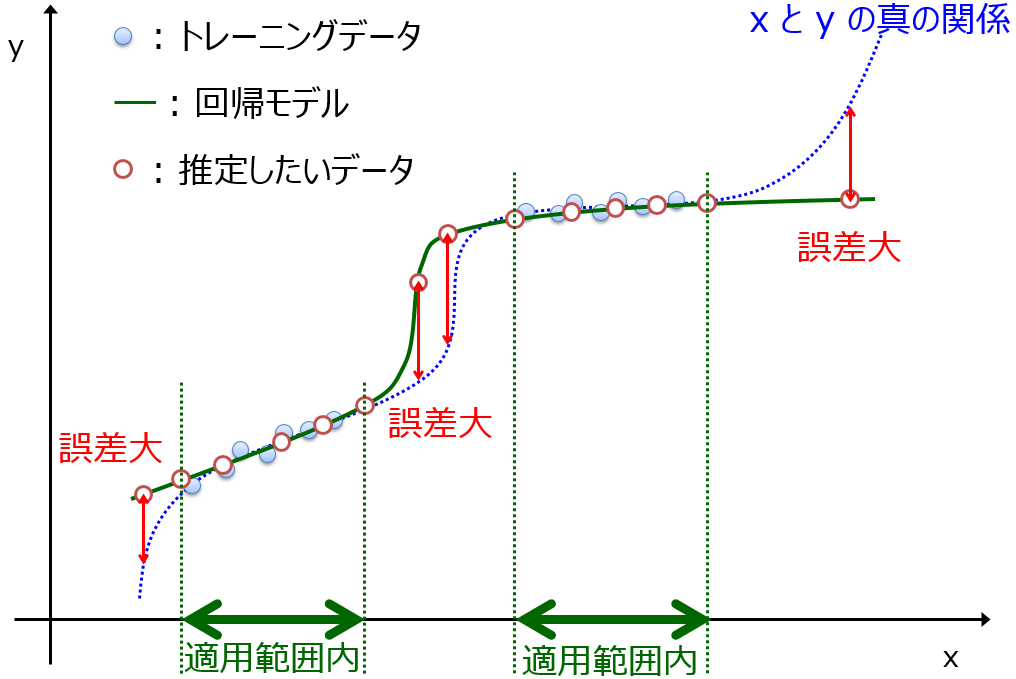
そこで考えました。y の値を推定したいサンプル郡がわかっているのであれば、そのサンプル郡が AD の中に入るようにモデルを構築すればよいのでは!? と。変数選択に着目しまして、なるべく多くのサンプルが AD の中に入るように、変数選択しようと考えました。さらに、低次元化したほうがより AD が広がると考え、変数選択後に主成分分析 (Principal Component Analysis, PCA) で次元削減しました。PCA の詳細についてはこちら。
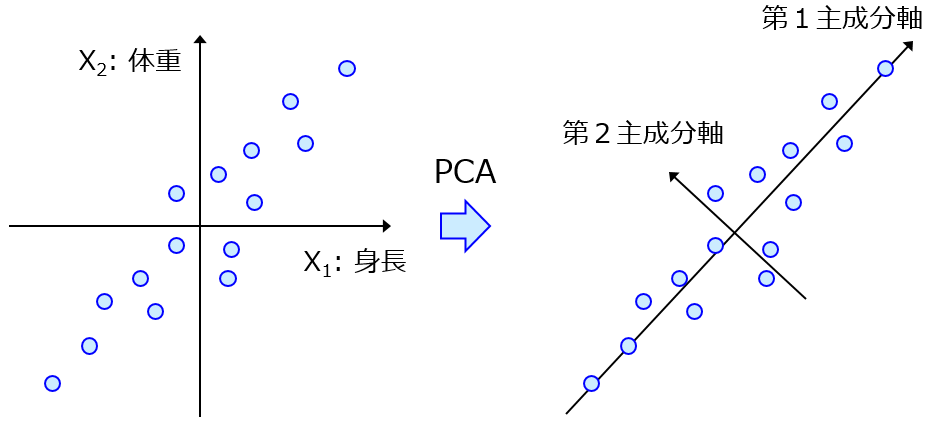
具体的に何をしたかというと、回帰分析において、遺伝的アルゴリズム (Genetic Algorithm, GA) を使った説明変数選択手法であるGenetic Algorithm-based Partial Least Squares (GAPLS) の適合度を少し変えました。GAPLS の詳細についてはこちら。

一般的な GAPLS では、各染色体の適合度を部分的最小二乗法 (Partial Least Squares, PLS) でクロスバリデーションしたときの r2 の値とします。クロスバリデーションで推定性能を評価できるとして、推定性能が高くなるように GA で変数を選択するわけです。
推定性能だけでなく AD も考えるため、適合度を次のように変えます。
適合度 = クロスバリデーション後の r2 + w × (AD 内のサンプル数)
w は重みです。染色体から選択された変数のみを用いて、モデル構築用データと新しいデータを合わせて PCA で低次元化したのちに、AD 内のサンプル数を数えます。AD は k 最近傍法で設定します。また PLS でクロスバリデーション後の r2 を計算します。これにより上の適合度を計算できるわけです。
w をいろいろ変えて検討したところ、クロスバリデーション後の r2 が変数選択前より向上し、y を推定したいすべてのサンプルが AD 内に入るような変数を選択できました。
ここまではよかったのですが、いざ AD 内に入った新しいサンプル郡における y の値を推定してみると、推定性能は向上しませんでした。変数選択することでローカルな AD は変えられますが、universal AD も大事で、それは変わらないわけです。Universal AD についてはこちら。

変数選択しても結局、universal AD の外のデータは、universal AD の外であるため、推定誤差は小さくならなかったのだろうと考えています。また、ある指標を評価に使えることと、その指標を最適化することでうまくいくことは違うのだろうと考えています。
以上です。
質問やコメントなどありましたら、twitter, facebook, メールなどでご連絡いただけるとうれしいです。

